
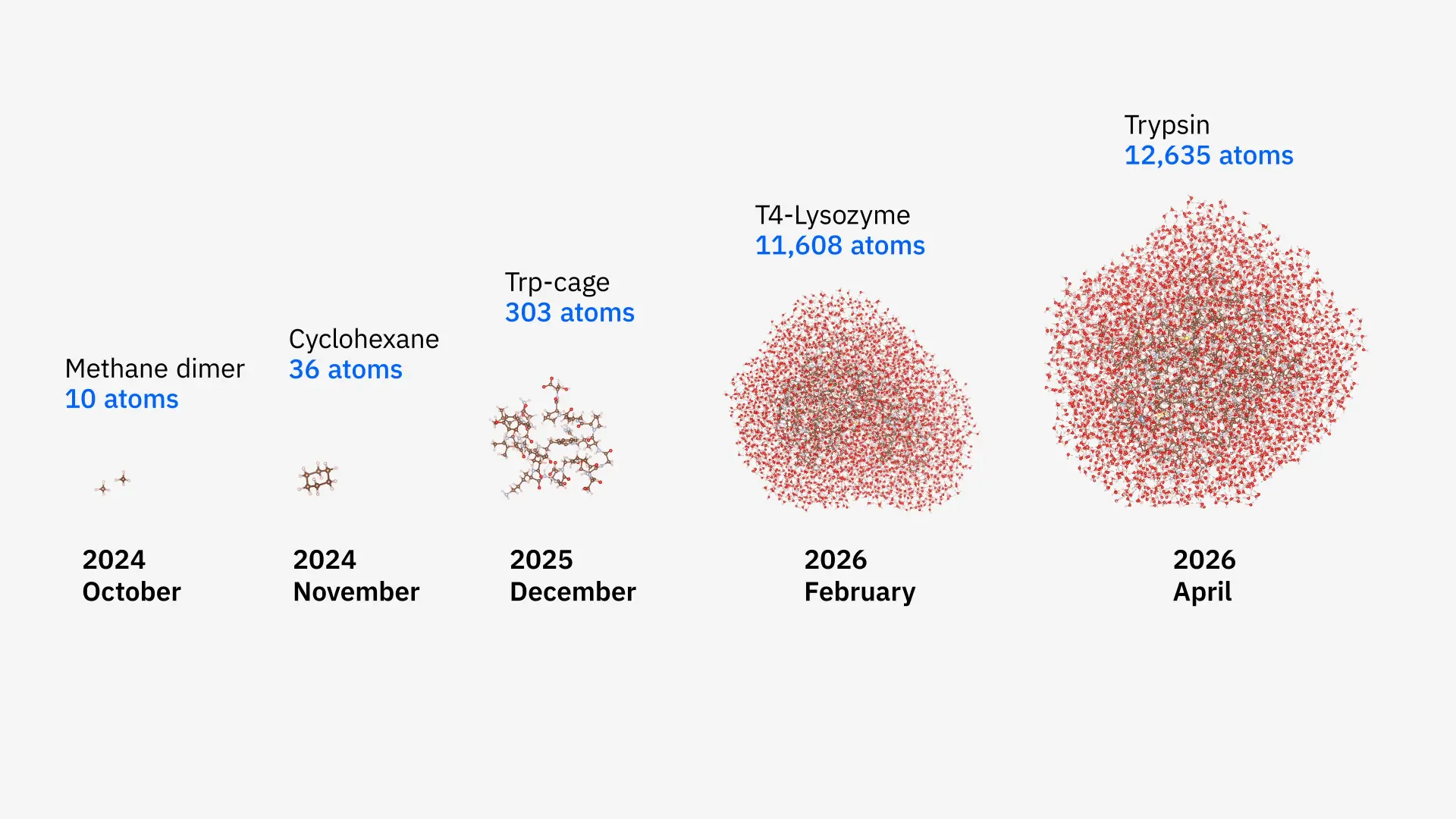
A collaborative team from Cleveland Clinic, RIKEN, and IBM has successfully simulated a protein-ligand complex spanning 12,635 atoms, marking a significant milestone in quantum-centric supercomputing. This simulation, which modeled the proteins T4-Lysozyme and Trypsin in a liquid water solution, represents the largest biologically meaningful molecular simulation performed on quantum hardware to date. The achievement demonstrates a 40-fold increase in system size and a 210-fold improvement in accuracy compared to similar benchmarks conducted only six months prior, signaling that quantum computers are maturing into practical scientific tools for the life sciences.
The technical breakthrough was driven by a novel hybrid algorithm called EWF-TrimSQD (Embedding Wave Function – Trimmed Sample-based Quantum Diagonalization). This framework utilizes “quantum-centric supercomputing,” where classical computers deconstruct massive protein-ligand complexes into smaller, computable “fragments.” The most complex, entangled clusters are then offloaded to IBM Quantum Heron processors. In this study, up to 94 qubits performed nearly 6,000 quantum operations per simulation to calculate the electronic structures of the fragments. These results were then reassembled by classical supercomputers—specifically RIKEN’s Fugaku and the University of Tokyo’s Miyabi-G—to provide a complete representation of the molecule.
The transition from the previous 303-atom “Trp-cage” benchmark to over 12,000 atoms was made possible by optimizing the classical “fragment bath” selection. Researchers implemented linear-scaling methods which restrict the quantum-mechanical calculations to a local sphere of 7–10 angstroms around specific atoms, as electronic entanglement typically diminishes at greater distances. This refinement prevented the classical overhead from scaling exponentially, making the simulation of Trypsin—a protein essential for human digestion—computationally feasible. The team collected 1.3 billion measurement outcomes across more than 100 hours of execution, the most resource-intensive quantum chemistry task reported to date.
This work addresses a fundamental bottleneck in drug discovery: accurately predicting how a drug candidate binds to a target protein. Existing classical computational methods struggle with the complexity of these interactions as molecules grow in size, often leading to decade-long development timelines and high failure rates in clinical trials. By providing a scalable framework for modeling atomic movement and computing accurate molecular energies, quantum-centric supercomputing offers a path toward significantly shorter pharmaceutical development cycles. The ability to model proteins in a realistic water solution, rather than in isolation, further increases the scientific relevance of these virtual models for real-world drug design.
Looking ahead, the researchers envision these methods evolving toward simulating complex enzyme catalysts and drug mechanisms that currently require exhaustive physical experimentation. Lead author Dr. Kenneth Merz of Cleveland Clinic noted that the current trajectory suggests quantum computing may soon surpass the best purely classical approaches in chemical accuracy. As hardware continues to advance—including the anticipated 2029 release of the fault-tolerant IBM Quantum Starling—the integration of quantum processors into global supercomputing networks is expected to redefine the boundaries of computational biology and materials science.
You can find the official press release from IBM here, the technical summary on the IBM Research Blog here, the institutional perspective from Cleveland Clinic here, and the preprint study on arXiv here.
May 5, 2026
