
Lockheed Martin and IBM have applied sample-based quantum diagonalization (SQD) to simulate the electronic structure of methylene (CH₂), marking the first use of SQD for an open-shell molecule. The collaboration highlights SQD as a strong candidate for near-term quantum advantage, capable of modeling strongly correlated systems that challenge classical computational chemistry.
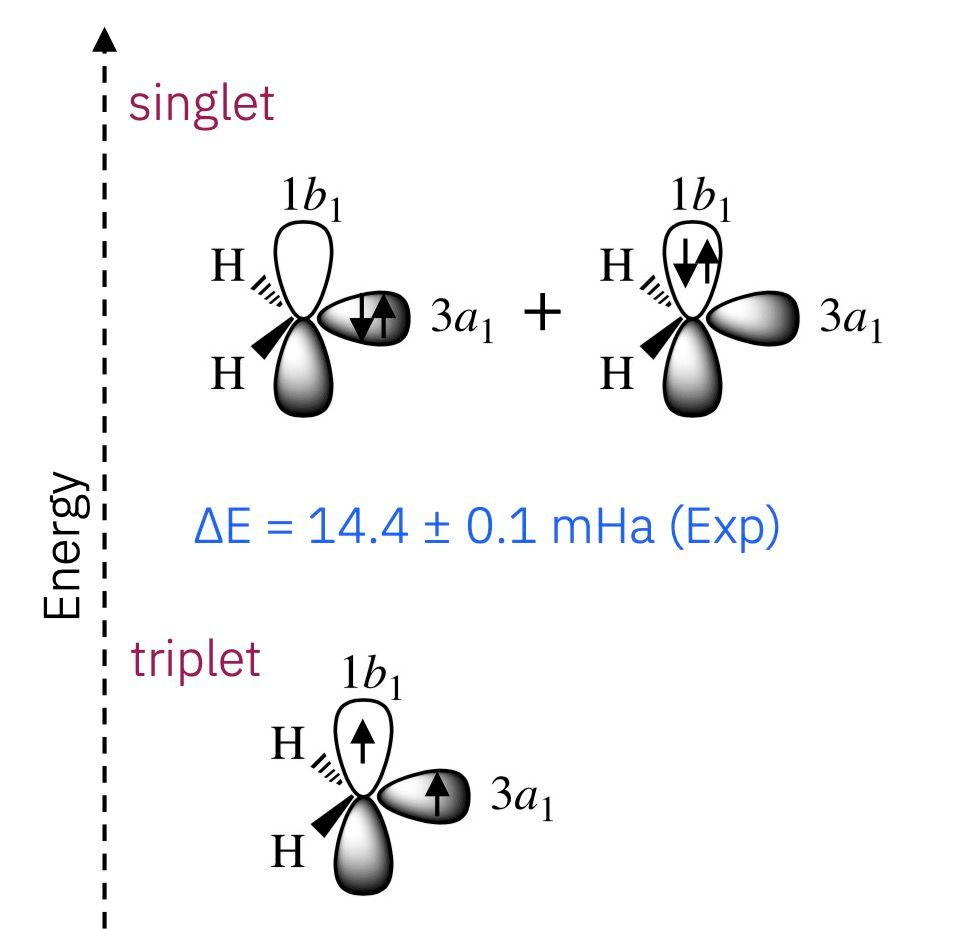
In this study, the team simulated singlet and triplet states of CH₂, an open-shell diradical relevant to combustion and atmospheric chemistry. Using IBM’s 52-qubit quantum processor within a hybrid quantum-centric supercomputing environment, researchers executed up to 3,000 two-qubit gates per experiment. The SQD calculations yielded accurate dissociation energies and singlet–triplet gaps, with results aligning closely with classical Selected Configuration Interaction (SCI) benchmarks.
Open-shell molecules such as CH₂ exhibit unpaired electrons and complex electronic correlations, which are difficult to model classically due to high resource requirements. By avoiding full wavefunction reconstruction and relying on sampled expectation values, the SQD approach reduces computational overhead while leveraging quantum-native electron entanglement.
This proof-of-concept study demonstrates that quantum simulations can address real chemical systems beyond idealized cases. The accurate prediction of CH₂’s electronic transitions lays groundwork for future quantum-enabled modeling in areas such as combustion reaction dynamics, molecular sensing, and aerospace materials design.
Read more in IBM’s blog post here and in the full research paper published in The Journal of Chemical Theory and Computation here.
May 29, 2025
